
On 26th May 2022 the In Vitro Diagnostic Regulation (IVDR, EU 2017/746) will become applicable. This regulation describes the rules for market access of in vitro medical devices in the European Economic Area. In this article, MedTech specialists Lisanne Karbaat and Tanja Hamacher at Holland Innovative cover what the new IVDR means for companies involved in the development and manufacture of medical devices.
In vitro diagnostic medical devices (IVDs) are a special category of medical devices which examine specimens from the human body in an external device outside the human body to acquire information about someone’s health status.

Figure 1: Example of in vitro diagnostic medical device
To date, these in IVDS were managed by the In Vitro Medical Devices Directive (IVDD). The new regulation is more stringent and aims at increasing the effectiveness and safety of in vitro medical devices.
If you have an IVDD self-declared product, you may remain on the market for a certain period of time (depending on the risk of your device) with a declaration of conformity, dated prior to the 26th of May 2022. If you have an IVDD certificate from a notified body, you may remain on the market until the expiration date of your certificate provided you do not make significant changes to your product. After the expiration of your certificate, you have to apply for an IVDR certificate.
Note that for ALL IVD manufacturers, the IVDR requirements regarding post-market surveillance, vigilance, and economic operators are directly applicable, as of the 26th of May 2022.
One of the major changes is the classification of IVDs. Under the IVDR, IVDs are divided into four classes A-D, with A the lowest risk class and D the highest. The seven classification rules take the test purpose, disease propagation risk, as well as the disease and specimen type into account. Classes B, C, and D must be assessed by a Notified Body. As a result, many devices will be reclassified to higher-risk classes. In the Netherlands, the percentage of devices that must be checked by a Notified Bodies increases from 7% under the IVDD to 84% under the IVDR (figure 2) (A. van Drongen et al., 2018, Rijksinstituut voor Volksgezondheid en Mileu). With our partners Panton and Fris & Fruitig, we are currently developing a new tool to determine the class of your IVD for our website CE Tool | Home.
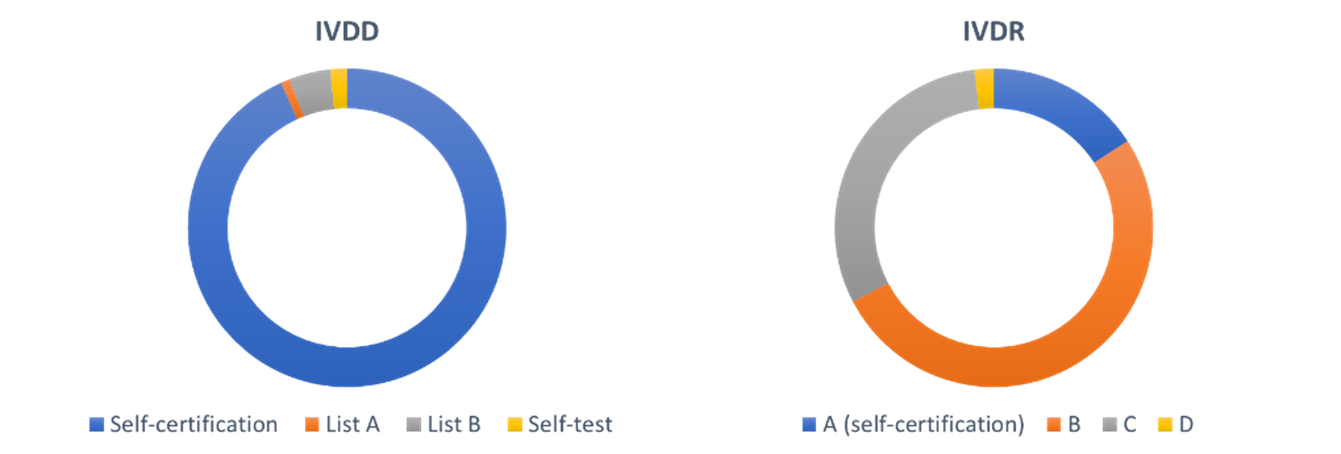
Figure 2: Percentage of devices classified in the Netherlands under the IVDD and IVDR. The number of devices checked by Notified Bodies increases from 7% to 84%. (A. van Drongen et al., 2018, Rijksinstituut voor Volksgezondheid en Mileu)
Other important requirements of the IVDR are generating, collecting, and assessing data to verify safety and performance prior- and post-market release, exchanging information in the European Database on Medical Devices (EUDAMED), and having a risk management system.
More info about the IVDR can be found here.
Do not hesitate to contact us if you have any questions or if you need any support for implementing the IVDR. The MedTech Team of Holland Innovative can help you with setting up and implementing regulatory requirements and technical files according to the IVDR.




.jpg?width=200&name=Holland%20Innovative%20summer%20academy%20-%20Project%20Management%20Masterclass%202%20(2).jpg)